



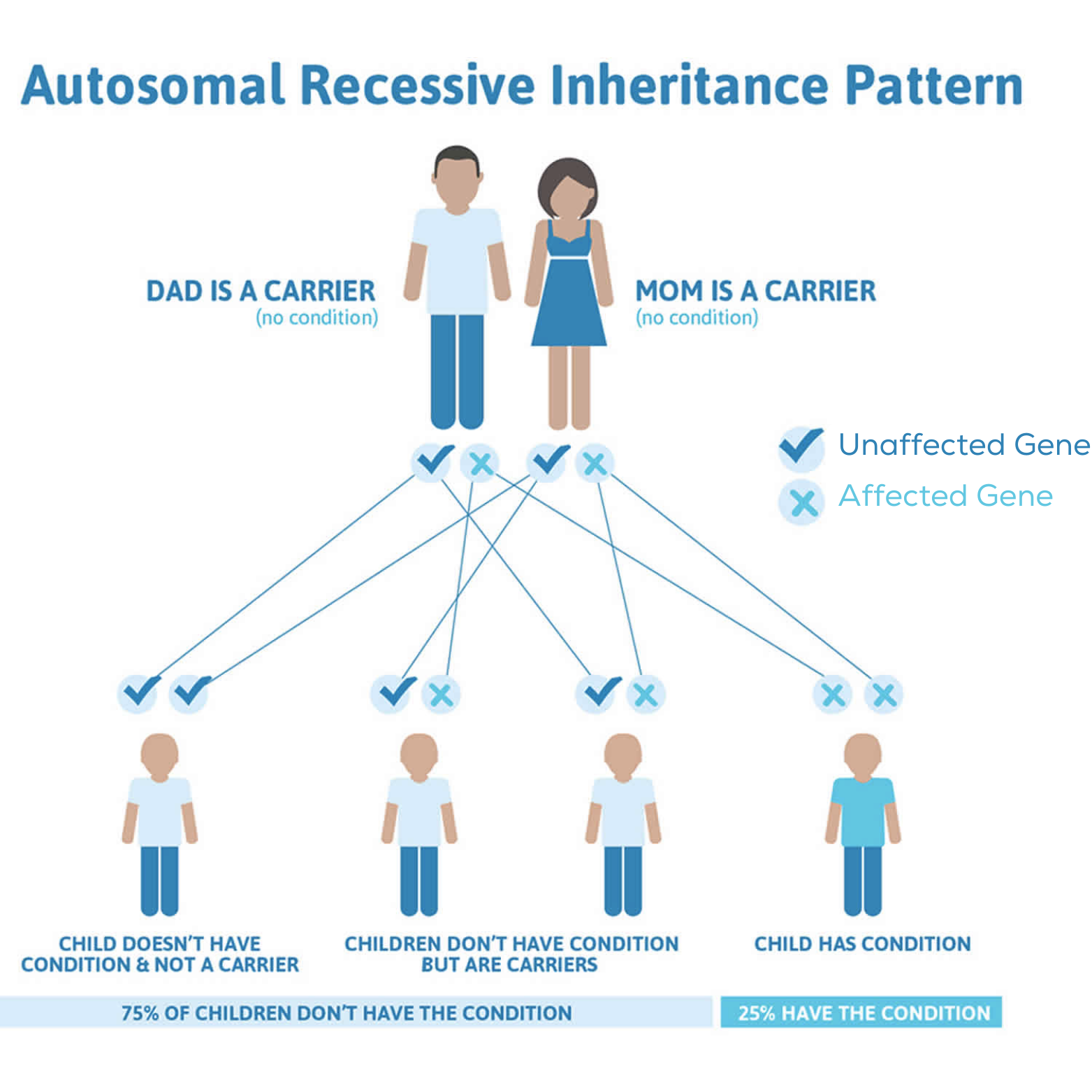
PH1
AGXT-
-Genmutation

AGXT-
-Enzymmangel
PH2
GRHPR-
-Genmutation

GR/HPR-
-Enzymmangel
PH3
HOGA1-
-Genmutation

HOGA1-
-Enzym-Mangel


OHF
Stiftung für Oxalose und Hyperoxalurie
AGXT-
-Genmutation
AGXT-
-Enzymmangel
GRHPR-
-Genmutation
GR/HPR-
-Enzymmangel
HOGA1-
-Genmutation
HOGA1-
-Enzym-Mangel