



PH1
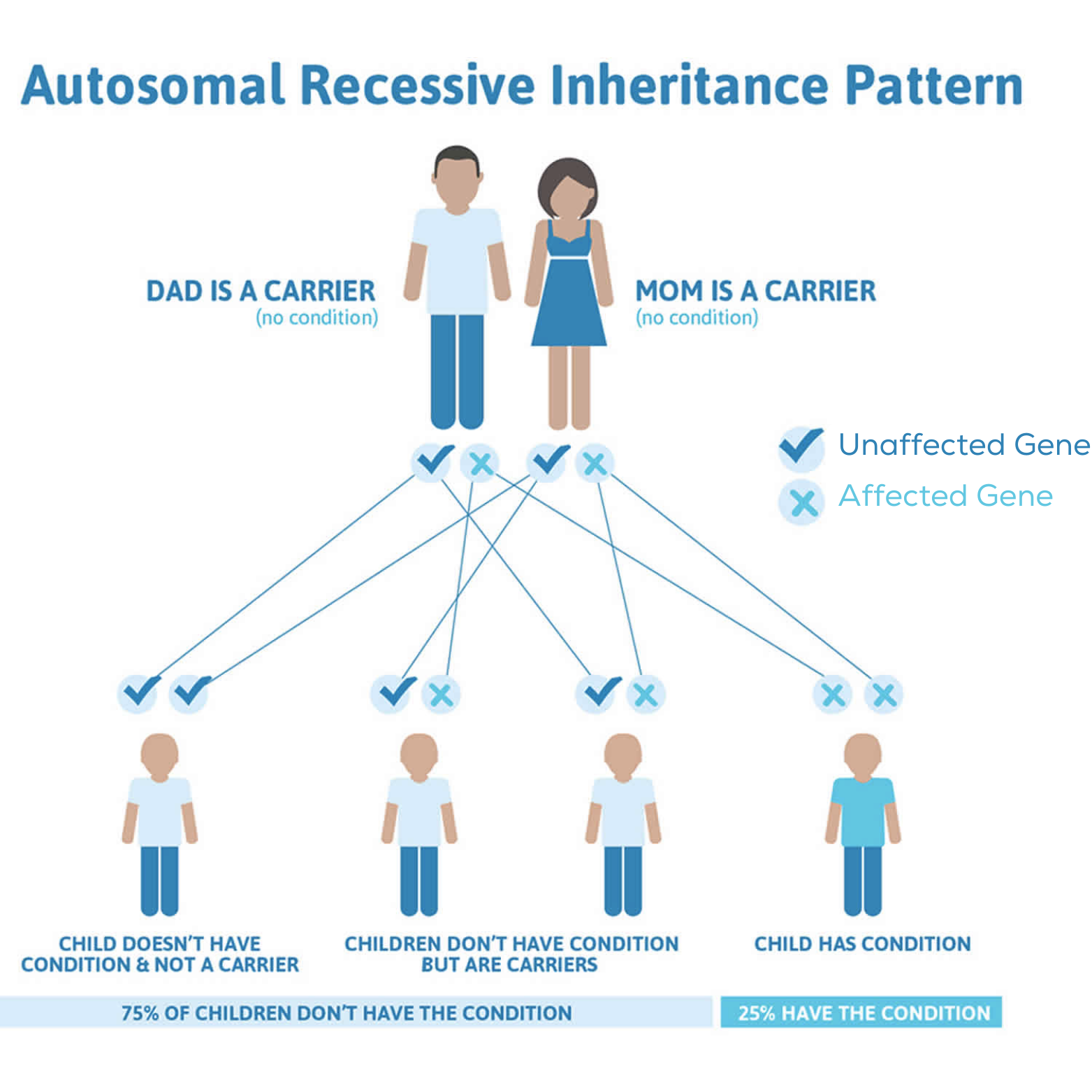
Mutación genética del gen AGXT (
)

AGXT
: deficiencia enzimática
PH2
Mutación genética del gen GRHPR (
)

Deficiencia de la enzima GR/HPR
PH3
Mutación genética del gen HOGA1

Deficiencia de la enzima HOGA1 (
)


OHF
Fundación para la Oxalosis y la Hiperoxaluria
Mutación genética del gen AGXT (
)
AGXT
: deficiencia enzimática
Mutación genética del gen GRHPR (
)
Deficiencia de la enzima GR/HPR
Mutación genética del gen HOGA1
Deficiencia de la enzima HOGA1 (
)