



PH1
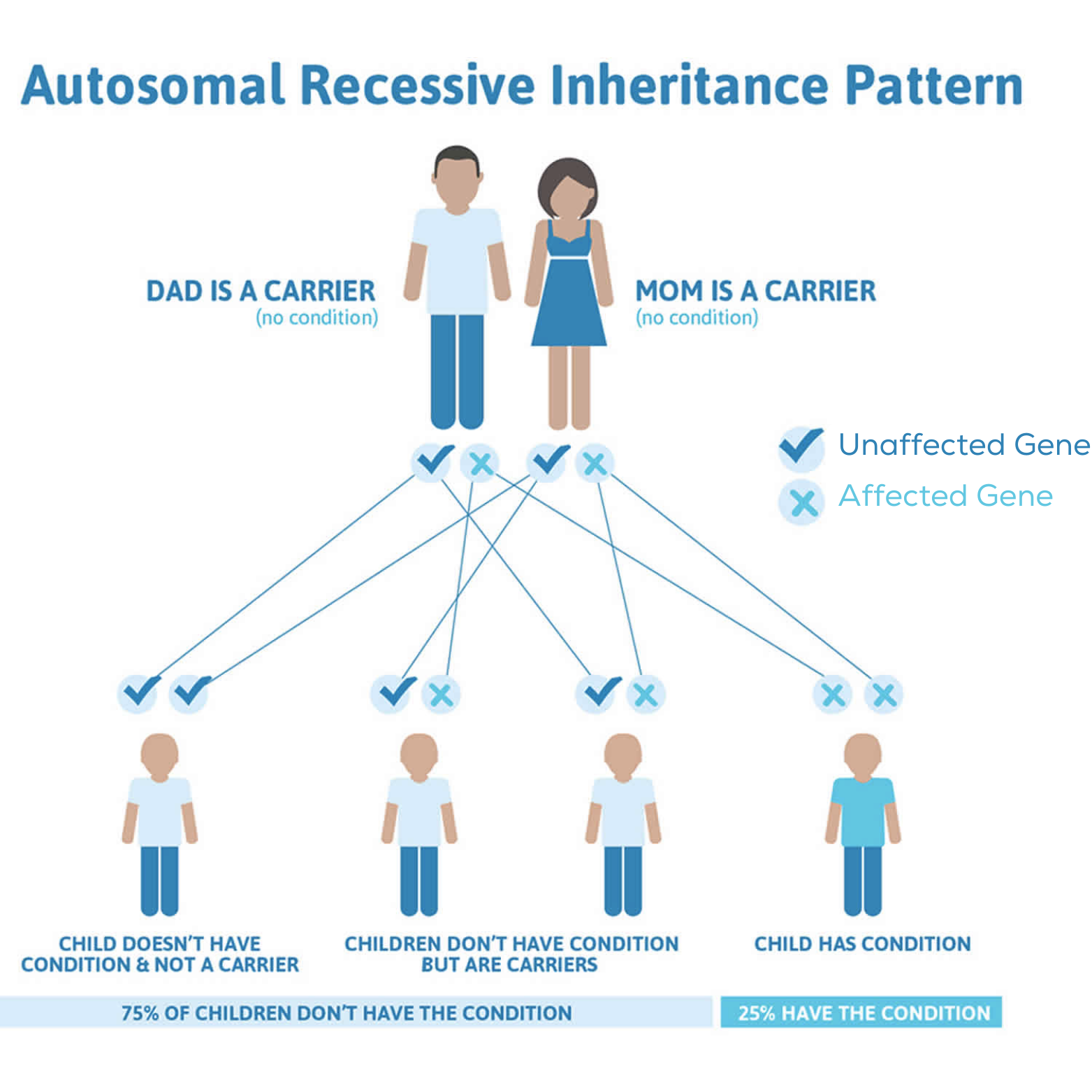
Mutation génétique de l'
e AGXT

Déficit en AGXT (
e)
PH2
Mutation génétique du gène GRHPR (
)

Déficit en enzyme GR/HPR (
)
PH3
Mutation génétique du gène HOGA1 (
)

Déficit en enzyme HOGA1 (
)


OHF
Fondation pour l'oxalose et l'hyperoxalurie
Mutation génétique de l'
e AGXT
Déficit en AGXT (
e)
Mutation génétique du gène GRHPR (
)
Déficit en enzyme GR/HPR (
)
Mutation génétique du gène HOGA1 (
)
Déficit en enzyme HOGA1 (
)